



According to the National Medicines Regulatory Authority Act No. 05 of 2015 the National Medicines Regulatory Authority shall be responsible for the regulation and control of registration, licensing, manufacture, importation and all other aspects pertaining to medical devices in a manner compatible with the National Medicines Policy ;
Definition of a medical device :
As defined in the Act a “medical device” means any instrument, apparatus, appliance, software, material or any other article, whether used single or in combination, including the software necessary for its proper application intended by the manufacturer used in or on human beings for the purpose of :
- Diagnosis, prevention, monitoring, treatment or alleviation of disease ;
- Diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap ;
- Investigation, replacement or modification of the anatomy or of a physiological process ;
- Control of conception ;
and which does not achieve its intended action in or on the human body by pharmacological, immunological or metabolic means but which may be assisted in its function by such means ;
A medical device does not include an Ayurveda device or a Homeopathy device ;
Scope and Responsibilities of the NMRA in the approval of medical devices
The National Medicines Regulatory Authority (NMRA) is responsible to build a healthier nation by ensuring that medical devices available for supply in Sri Lanka are of acceptable quality, safety and fit for their intended purpose. As such all Medical devices which are categorized as in above definition and should be registered with the Authority and licence to be obtained for manufacturing, importation, re-packaging, sale, distribution and offered for sale in Sri Lanka.
All foreign medical device manufactures should submit application for registration through a Marketing Authorization Holder (local agent) in Sri Lanka who shall be responsible for the registration, licencing, importation, sale and distribution, handing of quality failures and all aspects pertaining to the particular medical device in Sri Lanka.
The Medical Device Evaluation Committee (MDEC) formed under NMRA Act carries out technical evaluation of the medical devices forwarded for registration by considering the quality, safety, effectiveness, need and cost of such devices. The MDEC consist of experts drawn from various specialties in medical and pharmaceutical fields who meets monthly to decide on applications submitted for marketing authorization of medicines and to make policy decisions relevant to marketing authorization of medicines.
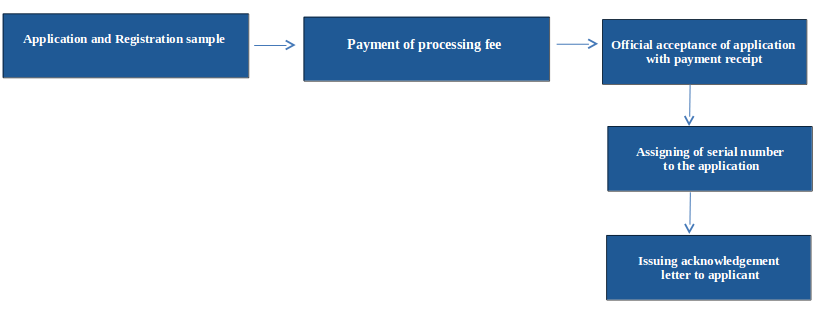
Flow chart of registration process of medical devices is as follows

Steps involved for registration of medical devices

Submission of registration application
Note : In addition approval of the manufacturing site will be initiated in future
Documents for obtaining sample import licence
- Fulfilled application Form C schedule IV of Cosmetics, Devices and Drugs Regulations (The gazette of the Democratic Socialist Republic of Sri Lanka (Extraordinary) No. 378/3 of 1985)
- A copy of business registration certificate of the applicant[should indicate details of the board of directors, secretarial board (Form 48 )]
- letter of authorization from the manufacturer appointing the Market Authorization Holder
- Copy Free Sale Certificate of particular product from relevant health authority of country of origin
- Price comparison and CIF price (Separate applications should be submitted for different manufactures)
Application for medical device registration
- The application for registration shall be made along with the required documents in Schedule I, Form A (link to forms) of Cosmetics, Devices and Drugs Regulations (The gazette of the Democratic Socialist Republic of Sri Lanka (Extraordinary) No. 378/3 of 1985)
- Documents should be in English, in a legible font size, printed in one side A4 and submitted in a hard file cover (Box file) and all pages should be numbered from top to bottom and vice versa with an index. Certified English translations should be submitted if the original certificates or licences issued in any other languages by relevant competent authorities
- The applications for registration are processed only if they are complete and as per specifications
- Separate applications should be made in respect of each device to be registered. [i.e. products containing different specifications, different brands] . Products of foreign manufacturers should be submitted through a Marketing authorization Holder
- A separate application is required for each product i.e. product containing different specifications or by a different manufacturer shall require a separate application for product registration
- Separate application to be submitted when a medical device consists of different constituents/ components. Each and every component of that system is registered separately.
- Orthopaedic system - separate applications should be produced for bone plates, nails, pins, screws
- Dental appliances
- A medical device although the manufacturing process is same and shares a common intended purpose is registered separately
- Condoms with different texture (flavour)
- Syringes with different volumes
- CV catheters, haemodialysis catheters, blood bags (for single, double and triple)
- In vitro diagnostic devices that consist of reagents or article intended to be used in combination to complete a specific intended purpose is registered as a group
- Haematology analyser with standards, programme and reagents Or as separately
- Blood grouping reagent, blood glucose monitoring system with component
- A medical device consisting a collection of devices and has a common intended purpose is registered as a group.
- Electro surgical unit with standard accessories (electrodes, electrode holders, leads, Plates, plug adopter)
- Anaesthesia machine with standard accessories
- Nebulizer system
General Documents/ Requirements
- Fulfilled Schedule I, Form A & Form B
- Copy of sample import licence
- Free sale certificate or certificate to foreign government issued from relevant health authority of country of origin and certified by Sri Lankan Embassy of country of origin or foreign affairs
- Fully packed samples (two) of devices in the form that is intended to be marketed (including Lot no., Man. Date, Exp.date, Manufacturer’s & Importers details and when required sufficient quantity for analysis)
- Letter of authorization from the manufacturer appointing the Market Authorization Holder
- List of countries which the device is approved or registered for sale with copies to prove registration status
Technical Documents
Following documents should be submitted in addition to the basic documents where necessary / if available
- Final product inspection report (for electro medical equipment and machines) and finished product test report for other products Submit relevant report issued by the manufacturer or third party laboratory for batch release of the product
- Test reports for below mentioned items
- Independent analytical certificates (original report) from Industrial Technology Institute (ITI), Sri Lanka or govt. accreted laboratory in of the country of origin for products which are directly in contact with the blood stream
Eg: disposable syringes, disposable needles, IV cannulas IV catheters, fistula needles etc
Test reports are to be submitted according to pharmacopeia standards where the standards are available - Analytical test reports from Sri Lanka Standard Institute (SLSI) for products feeding bottles, tooth brushes and medical gas cylinders
- Analytical test reports from Industrial Technology Institute (ITI)
Eg: plasters, gauze, sanitary napkins, bandages, latex condoms, surgical and examination gloves etc. - In addition reports from NMQAL may be requested
- Independent analytical certificates (original report) from Industrial Technology Institute (ITI), Sri Lanka or govt. accreted laboratory in of the country of origin for products which are directly in contact with the blood stream
- Material test report for sutures, medical instruments such as forceps, scissors etc.
- Certification for quality management system according to ISO 13485 from authorized notified body in order to access the design, development, manufacturing as well as for post marketing monitoring of safety and performance of the manufacturer
- CE accreditation from authorized notified body in order to prove the quality assurance system of the product and EC design examination certificate (if applicable)
- Stability data for entire shelf life of the finished products should be provided (if applicable)
- In addition following requirements should be fulfilled for Absorbable sutures with the application
- All the samples of absorbable sutures will be kept at the NMRA for six (6) months before sending for evaluation to the relevant consultant
- Details of the raw material sources, purchasing details should be provided
- Where necessary certificate of approval from relevant authorities should be provided
- For radiation emitting devices approval obtained from Atomic energy Authority of Sri Lanka
- Certification from the relevant health authority of the country of manufacturer that the product is free from BSE (Bovine Spongiform Encephalopathy) should be obtained for animal derived products
Eg : Surgical Catgut
- Biological evaluation/biocompatibility test report of medical device as per ISO standards (if applicable)
- Risk management analysis as per ISO standard (if applicable)
- Recently issued validation report for sterilization process for two commercial batches (if applicable)
Product Label (primary and secondary)
Submit original label including following information
- Name of product [Approved name and brand name (if any)]
- Name and address of the actual manufacturer
- Whether the product is sterile and mode of sterilization
- Storage conditions specifying the temperature
- Manufacturing date, Expiry date and Lot no/ batch no. (if applicable)
Patient information Leaflet
- Product information leaflet for the products which are individually handled by the patient in the household should be in both Sinhala and Tamil languages
- Eg : Glucometers, Hearing aids, spacer device etc.
- The Provisional Registration for a period of one year (or two) will be issued for first time registration and is specified in the certificate
- The Full Registration of a product is valid for a period of five years and is specified in the certificate
- When additional data are requested, the applicant will have to furnish additional information requested by the authority within 3 months to facilitate further evaluation
- If the product is rejected, the market authorization holder will be able to appeal for registration
- Application for renewal should be made before six months from the date of expiry of registration
- A grace period will extend until the decision is given to the application for renewal
- If the requirements for registration are not satisfactory the application will be rejected completely
- Any change in product name, product specifications, packaging, indications, contents of product label, package insert, or product literature, or any relevant particulars of the registered product should not be made without the prior approval of the authority
- The registration of the product maybe cancelled if changes are made without the prior approval of the authority
- Any change of product which affects quality, safety & efficacy of the product should require a new application for registration
- The applicant should be responsible for the product and all information supplied in support of his application for registration of the product
- The applicant should be responsible for updating any information relevant to the product/ application. The NMRA should be informed in a timely manner any change in product information during the course of evaluation, and after product registration, especially if the information pertaining to rejection/ withdrawal, additional data on product quality, effectiveness and safety or current Good Manufacturing Practice (cGMP) compliance of the manufacturers
- The applicant should provide additional documents for renewal of registration, six months before the expiry of certificate of registration
- The marketing authorization holder must assume responsibility for the quality, safety and effectiveness of his products